


治験の統括報告書の構成と内容に関するガイドライン
平成8年5月1日 薬審第335号
各都道府県衛生主管部(局)長あて 厚生省薬務局審査課長通知
STRUCTURE AND CONTENT OF CLINICAL STUDY REPORTS
Recommended for Adoption at Step 4 of the ICH Process on 30 November 1995 by the ICH Steering Committee
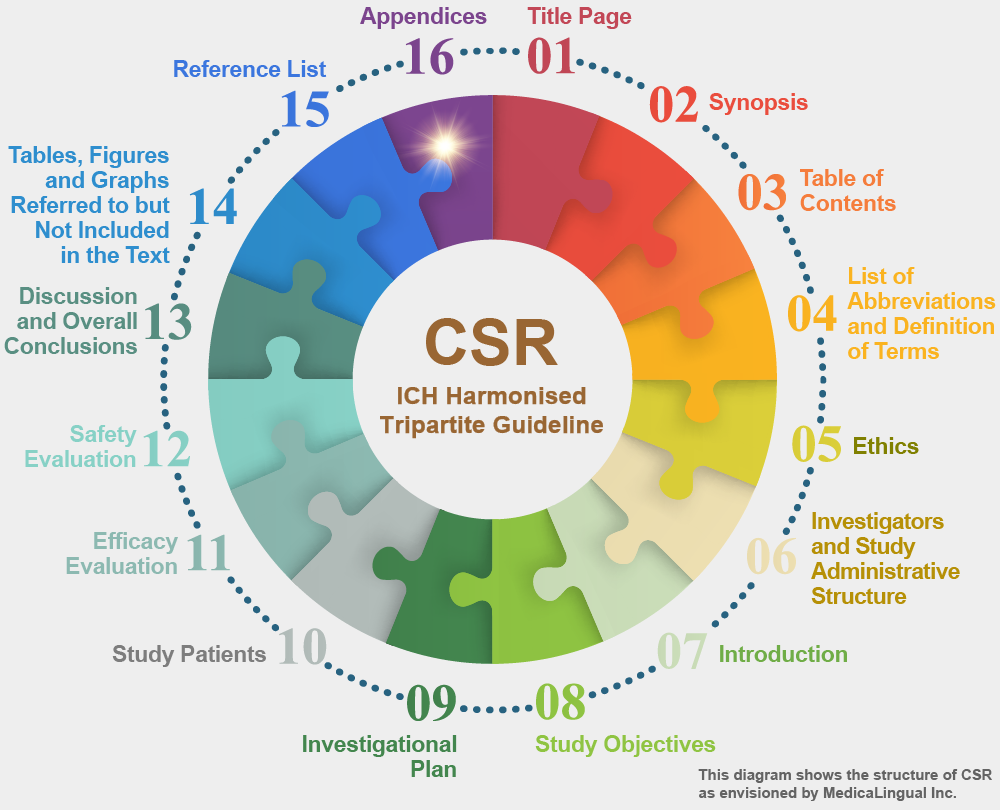
16. 付録
この章の冒頭には,総括報告書において利用可能な全ての付録の完全な一覧表をつけること。
16. APPENDICES
This section should be prefaced by a full list of all appendices available for the study report.
Where permitted by the regulatory authority, some of the following appendices need not be submitted with the report but need to be provided only on request.
The applicant should therefore clearly indicate those appendices that are submitted with the report.
N.B. In order to have appendices available on request, they should be finalised by the time of filing of the submission.
16.1 治験に関する情報
16.1.1 治験実施計画書及びその改訂
16.1.2 症例記録用紙の見本(内容の異なるページのみ)
16.1.3 治験審査委員会の一覧(確認が行われた年月日,並びに委員の氏名及び職名),患者への説明文書及び同意書の見本
16.1.4 治験責任医師及び他の重要な治験参加者の一覧表及び説明(簡潔な(1ぺージ)履歴書又は治験の実施に関連する訓練や経験についての履歴書と同等の要約を含む)
16.1.5 治験総括(調整)医師又は治験依頼者の医学責任者の署名
16.1.6 複数ロットが用いられた場合には,治験に用いられたロットごとの薬剤を投与された患者一覧表
16.1.7 無作為化の方法及びコード(患者の識別及び割り付けられた治療)
16.1.8 監査手順に関する資料,監査証明書(可能であれば)
16.1.9 統計手法に関する文書
16.1.10 臨床検査に関して施設間の標準化及び品質保証を行ったのであればその方法と手順に関する文書
16.1.11 治験に基づく公表文献
16.1.12 総括報告書で引用された重要な公表文献
16.1 STUDY INFORMATION
16.1.1 Protocol and protocol amendments
16.1.2 Sample case report form (unique pages only)
16.1.3 List of IECs or IRBs (plus the name of the committee Chair if required by the regulatory authority) – Representative written information for patient and sample consent forms
16.1.4 List and description of investigators and other important participants in the study, including brief (1 page) CVs or equivalent summaries of training and experience relevant to the performance of the clinical study
16.1.5 Signatures of principal or coordinating investigator(s) or sponsor’s responsible medical officer, depending on the regulatory authority’s requirement
16.1.6 Listing of patients receiving test drug(s)/investigational product(s) from specific batches, where more than one batch was used
16.1.7 Randomisation scheme and codes (patient identification and treatment assigned)
16.1.8 Audit certificates (if available) (see Annex IVa and IVb of the guideline)
16.1.9 Documentation of statistical methods
16.1.10 Documentation of inter-laboratory standardisation methods and quality assurance procedures if used
16.1.11 Publications based on the study
16.1.12 Important publications referenced in the report
16.2 患者データ一覧表
16.2.1 中止症例
16.2.2 治験実施計画から逸脱した症例
16.2.3 有効性の解析から除外された症例
16.2.4 人口統計学的データ
16.2.5 服薬遵守及び(又は)薬物濃度データ(可能であれば)
16.2.6 個々の有効性反応データ
16.2.7 患者ごとの有害事象一覧表
16.2. PATIENT DATA LISTINGS
16.2.1 Discontinued patients
16.2.2 Protocol deviations
16.2.3 Patients excluded from the efficacy analysis
16.2.4 Demographic data
16.2.5 Compliance and/or drug concentration data (if available)
16.2.6 Individual efficacy response data
16.2.7 Adverse event listings (each patient)
16.2.8. Listing of individual laboratory measurements by patient, when required by regulatory authorities
16.3 症例記録
16.3.1 死亡,その他の重篤な有害事象発現例及び有害事象による投与中止例の症例記録
16.3.2 提出された他の症例記録
16.3 CASE REPORT FORMS
16.3.1 CRFs for deaths, other serious adverse events and withdrawals for AE
16.3.2 Other CRFs submitted





