


医薬審発第899号 CTD通知(別紙 4)
CTD 非臨床に関する文書の作成要領に関するガイドライン
(2002年9月11-12日ワシントン会議修正版)
ICH HARMONISED GUIDELINE – M4S –
(Numbering and Section Headers have been edited for consistency and use in e-CTD as agreed at the Washington DC Meeting, September 11-12, 2002)
一般原則
本ガイドラインは、非臨床試験の概括評価、概要文及び概要表を3極間で調和を勧めるために作成されたものである。
General Principles of Nonclinical Overview and Summaries
This guideline provides recommendations for the harmonisation of the Nonclinical Overview, Nonclinical Written Summary, and Nonclinical Tabulated Summaries.
非臨床試験の概要文及び概要表の作成の主要な目的は非臨床試験について事実に基づく包括的な概要を示すことである。概括評価には、成績の説明、得られた成績の臨床との関連性、医薬品の品質情報との関連づけ及び医薬品の安全使用に関連する非臨床試験との関連性(すなわち、添付文書等に記載すべき事柄)について記載する。
The primary purpose of the Nonclinical Written and Tabulated Summaries should be to provide a comprehensive factual synopsis of the nonclinical data. The interpretation of the data, the clinical relevance of the findings, cross-linking with the quality aspects of the pharmaceutical, and the implications of the nonclinical findings for the safe use of the pharmaceutical (i.e., as applicable to labeling) should be addressed in the Overview.
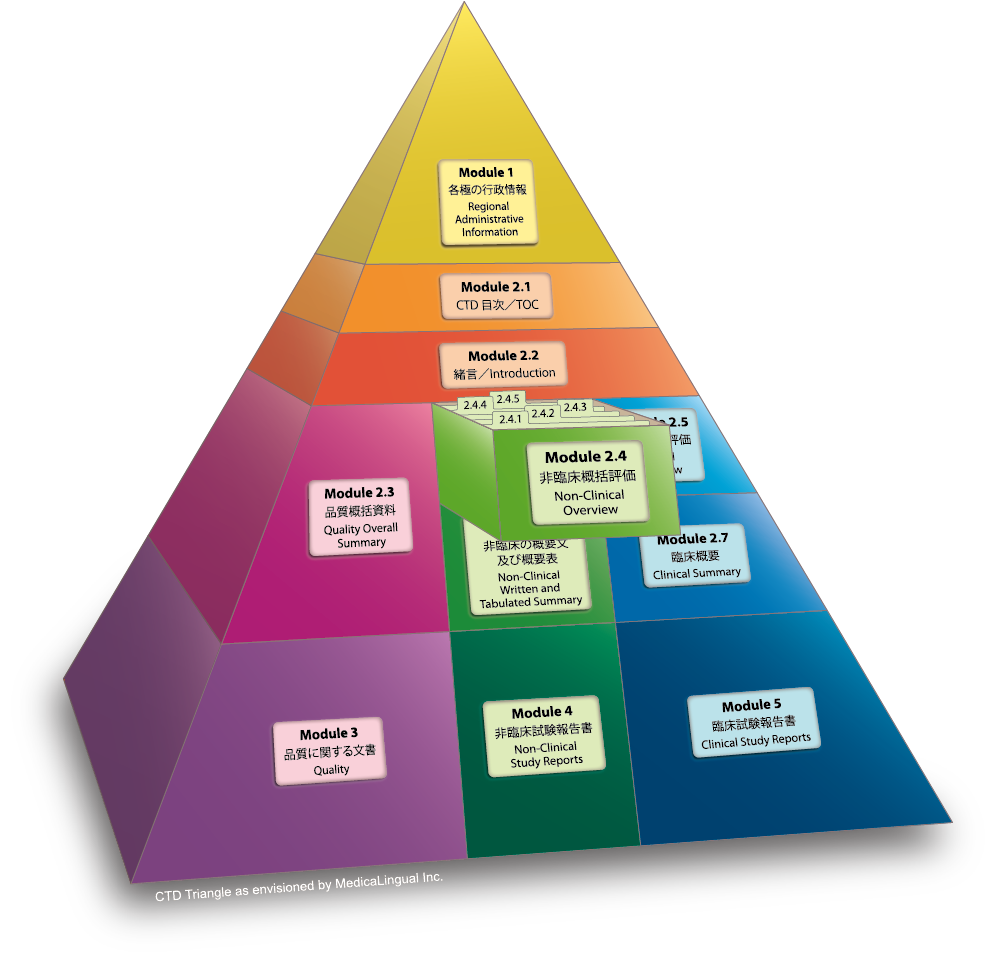
2.4 非臨床試験の概括評価
本章では、CTDにおける情報の全体像を示すこと。原則として、非臨床試験の概括評価は概ね30頁を超えないこと。
2.4 NONCLINICAL OVERVIEW
The Nonclinical Overview should provide an integrated overall analysis of the information in the Common Technical Document. In general, the Nonclinical Overview should not exceed about 30 pages.
一般的情報
非臨床試験の概括評価では、医薬品の薬理評価、薬物動態評価及び毒性評価について包括的で客観的な評価結果を示すこと。試験の実施に関連するガイドラインがある場合には、ガイドラインを考慮するとともに、ガイドラインからの逸脱については、考察しその妥当性を示すこと。非臨床試験の計画についても、考察し妥当性を示すこと。提出された試験がGLPに適合していることについて示し、必要に応じて、非臨床試験で得られた所見と医薬品の品質、臨床試験の結果及び類薬で認められた作用との関連を示すこと。
General Aspects
The Nonclinical Overview should present an integrated and critical assessment of the pharmacologic, pharmacokinetic, and toxicologic evaluation of the pharmaceutical. Where relevant guidelines on the conduct of studies exist, these should be taken into consideration, and any deviation from these guidelines should be discussed and justified. The nonclinical testing strategy should be discussed and justified. There should be comment on the GLP status of the studies submitted. Any association between nonclinical findings and the quality characteristics of the human pharmaceutical, the results of clinical trials, or effects seen with related products should be indicated, as appropriate.
バイオテクノロジー応用医薬品を除き、原薬及び製剤中に含まれる不純物と分解物について評価し、推測される薬理及び毒性作用に関して判明している点についても示すこと。この際、原薬及び製剤について定められた不純物の上限が適切であることを示し、品質に関する資料との間で相互参照できるように記載すること。また、非臨床試験で用いた化合物と市販予定製品の間の光学活性、化学形態及び不純物プロフィールの相違について示すこと。バイオテクノロジー応用医薬品については非臨床試験で用いられた物質、臨床試験で用いられた物質及び市販予定製品である物質の同等性の評価結果を示すこと。新添加剤*を含む製剤の場合は、その安全性に関する情報を示すこと。
*使用前例のない,又は使用前例があっても投与経路が異なる若しくは前例を上回る量を使用する場合(邦訳時追記)
Except for biotechnology-derived products, an assessment of the impurities and degradants present in the drug substance and product should be included along with what is known of their potential pharmacologic and toxicologic effects. This assessment should form part of the justification for proposed impurity limits in the drug substance and product, and be appropriately cross-referenced to the quality documentation. The implications of any differences in the chirality, chemical form, and impurity profile between the compound used in the nonclinical studies and the product to be marketed should be discussed. For biotechnology-derived products, comparability of material used in nonclinical studies, clinical studies, and proposed for marketing should be assessed. If a drug product includes a novel excipient, an assessment of the information regarding its safety should be provided.
関連する科学論文及び類薬の特性を考慮すること。申請者が試験を実施する代わりとして、公表科学論文を引用する場合には、試験計画及び現行のガイドラインからの逸脱について考察し、妥当性を示すこと。また、これらの引用された試験で使用された原薬のバッチの品質に関する情報の入手可能性についても示すこと。
非臨床試験の概括評価において、概要表を引用する場合は表X.X, 試験/報告書番号とすること。
Relevant scientific literature and the properties of related products should be taken into account. If detailed references to published scientific literature are to be used in place of studies conducted by the applicant, this should be supported by an appropriate justification that reviews the design of the studies and any deviations from available guidelines. In addition, the availability of information on the quality of batches of drug substance used in these referenced studies should be discussed.
The Nonclinical Overview should contain appropriate reference citations to the Tabulated Summaries, in the following format: (Table X.X, Study/Report Number).
内容及び構成様式
非臨床試験の概括評価は以下の順で示すこと:
- 非臨床試験計画概略
- 薬理試験
- 薬物動態試験
- 毒性試験
- 総括及び結論
- 参考文献一覧
薬理作用、作用機序及び予測される副作用を確認するために行なった試験を評価し、得られた結果の意義について考察すること。
薬物動態、トキシコキネティクス及び代謝データの評価においては、用いた分析法、薬物動態モデル及び得たパラメータの妥当性について考察すること。
薬理試験及び毒性試験での問題点をより詳細に検討するためには相互に参照することが適切である(病態に対する影響、生理学的変化、成分に対する抗体産生、トキシコキネティクスデータの動物種差の考察等)。
データ間の矛盾についても考察すること。代謝に関して動物種間の比較及び動物並びにヒトにおける全身曝露状態(AUC、Cmax及びその他の適切なパラメータ)の違いについて比較考察し、ヒトで考えられる副作用を予測するための非臨床試験の有用性及び限界を明らかにすること。
毒性の発現時期、程度(強さ)並びに持続期間、用量依存性並びに可逆性の程度(又は非可逆性)及び種差又は性差について評価し、重要な特徴について、特に以下の点に関して考察すること。
- 薬理作用
- 毒性変化
- 死亡原因
- 病理所見
- 遺伝毒性:化合物の化学構造、作用機序及び既知の遺伝毒性を示す物質との関連
- 化合物の化学構造と関連したがん原性、既知のがん原性を示す物質との関連、遺伝毒性及び曝露データ
- ヒトに対する発がんリスク:疫学的データが入手できる場合には、それらを考慮すること。
- 受胎能、胚胎児発生、出生前及び出生後の毒性
- 新生児への影響*
- 妊娠前並びに妊娠期間及び授乳期間並びに出生児の発達期間中投与による影響
- 局所刺激性
- その他の毒性試験:特別な問題を解明するための試験
* FDAが発生神経毒性を評価するために挿入したものである。以下、本別紙において同じ。(邦訳時追記)
ある作用及び事象に関連する全てのデータがまとめられるように、毒性試験の評価を論理立てて配列すること。動物からヒトへのデータの外挿は、以下の項目に関連付けて考察すること。
- 動物種
- 動物数
- 投与経路
- 投与量
- 投与又は試験期間
- 毒性試験に用いた動物種における無毒性量(NOAEL)及び毒性量での全身曝露とヒトにおける最高臨床推奨用量での曝露と関連付け。この情報は要約した図表で示すことが望ましい。
- 非臨床試験で認められた被験物質の作用とヒトで予測された又は認められた作用と関連付け。
丸ごとの動物を用いた試験に代わる試験を行った場合には、その科学的妥当性を考察すること。
総括及び結論では、非臨床試験によって示された当該医薬品の特徴を明確に記載し、また、目的とする臨床使用における当該医薬品の安全性が裏付けられるように、論理的かつ十分に検討された結論を導くこと。非臨床試験(薬理、薬物動態及び毒性)の結果を踏まえて、その医薬品のヒトでの安全使用(すなわち、添付文書等に記載すべき事柄)について考察すること。
Content and Structural Format
The Nonclinical Overview should be presented in the following sequence:
- Overview of the nonclinical testing strategy
- Pharmacology
- Pharmacokinetics
- Toxicology
- Integrated overview and conclusions
- List of literature references
Studies conducted to establish the pharmacodynamic effects, the mode of action, and potential side effects should be evaluated and consideration should be given to the significance of any issues that arise.
The assessment of the pharmacokinetic, toxicokinetic, and metabolism data should address the relevance of the analytical methods used, the pharmacokinetic models, and the derived parameters.
It might be appropriate to cross-refer to more detailed consideration of certain issues within the pharmacology or toxicology studies (e.g. impact of the disease states, changes in physiology, anti-product antibodies, cross-species consideration of toxicokinetic data).
Inconsistencies in the data should be discussed. Inter-species comparisons of metabolism and systemic exposure comparisons in animals and humans (AUC, Cmax, and other appropriate parameters) should be discussed and the limitations and utility of the nonclinical studies for prediction of potential adverse effects in humans highlighted.
The onset, severity, and duration of the toxic effects, their dose-dependency and degree of reversibility (or irreversibility), and species- or gender-related differences should be evaluated and important features discussed, particularly with regard to:
- pharmacodynamics
- toxic signs
- causes of death
- pathologic findings
- genotoxic activity – the chemical structure of the compound, its mode of action, and its relationship to known genotoxic compounds
- carcinogenic potential in the context of the chemical structure of the compound, its relationship to known carcinogens, its genotoxic potential, and the exposure data
- the carcinogenic risk to humans – if epidemiologic data are available, they should be taken into account
- fertility, embryofetal development, pre-and post-natal toxicity
- studies in juvenile animals
- the consequences of use before and during pregnancy, during lactation, and during pediatric development
- local tolerance
- other toxicity studies/ studies to clarify special problems
The evaluation of toxicology studies should be arranged in a logical order so that all relevant data elucidating a certain effect / phenomenon are brought together. Extrapolation of the data from animals to humans should be considered in relation to:
- animal species used
- numbers of animals used
- routes of administration employed
- dosages used
- duration of treatment or of the study
- systemic exposures in the toxicology species at no observed adverse effect levels and at toxic doses, in relation to the exposures in humans at the maximum recommended human dose. Tables or figures summarising this information are recommended.
- the effect of the drug substance observed in nonclinical studies in relation to that expected or observed in humans
If alternatives to whole-animal experiments are employed, their scientific validity should be discussed.
The Integrated Overview and Conclusions should clearly define the characteristics of the human pharmaceutical as demonstrated by the nonclinical studies and arrive at logical, well-argued conclusions supporting the safety of the product for the intended clinical use. Taking the pharmacology, pharmacokinetics, and toxicology results into account, the implications of the nonclinical findings for the safe human use of the pharmaceutical should be discussed (i.e., as applicable to labeling).





