


薬食審査発0517 第1 号
平成25 年5 月17 日
ICH Harmonised Tripartite Guideline
Having reached Step 4 of the ICH Process at the ICH Steering Committee meeting on 15 November 2012, this Guideline is recommended for adoption to the three regulatory parties to ICH
定期的ベネフィット・リスク評価報告 (PBRER)
1. 緒言
本ガイドラインで提案する定期的ベネフィット・リスク評価報告(PBRER)は、日米EU 医薬品規制調和国際会議(ICH)参加国や参加地域において、販売後の医薬品(現在、追加的な試験を行っている承認済み医薬品を含む。)に関する定期的なベネフィット・リスク評価の報告の共通の基準となることを意図している。
本ガイドラインでは、PBRER の推奨される内容と様式を定め、その作成及び提出において考慮すべきポイントを概説する。
本ガイドラインで使用されている用語の定義は「用語集(添付資料A)」に記載されており、本文中初めて記載された場合にアスタリスク(*)が付けられている。
Periodic Benefit-Risk Evaluation Report (PBRER)
1. INTRODUCTION
The Periodic Benefit-Risk Evaluation Report (PBRER) described in this Guideline is intended to be a common standard for periodic benefit-risk evaluation reporting on marketed products (including approved drugs that are under further study) among the ICH regions.
This Guideline defines the recommended format and content of a PBRER and provides an outline of points to be considered in its preparation and submission.
Definitions of many technical terms used in the Guideline are included in a glossary (Appendix A); the first mention of a term in the Guideline is identified with an asterisk (*).
1.1 背景
新医薬品が販売承認を受ける際には、安全性及び有効性は、一般に限られた患者数のデータにて確認されており、その多くはランダム化試験の管理された状況で検討が行われる。そのため、高リスクのサブグループや他の医薬品投与が必要な合併症を有する患者が臨床試験から除外されていることや、長期投与のデータが限られていることが多い。さらに、臨床試験に参加する患者は、有害事象のエビデンスに関して詳細にモニタリングされる。日常の診療下では、モニタリングはそれほど集中的に行われなかったり、より広い範囲(年齢、合併症、併用薬、遺伝子異常など)の患者が治療を受けたり、さらには、臨床試験ではあまりにまれなために発現することがない事象(例:重度の肝障害)が観察されたりすることがある。したがって、これらの要因は、定期的に、重要な知見が得られた際には直ちに、累積データの全体的評価を実施することで、安全性、有効性1 及び有用性1に関する情報を医薬品のライフサイクルに渡り継続的に分析する必要性があるという根拠となっている。新しい情報の大部分は安全性関連事項であるが、有用性、使用方法の制限、治療の選択肢、治療における医薬品の位置付けに関するその他多くの側面もベネフィット・リスク評価に関連することがある。
1 有効性(efficacy)及び有用性(effectiveness)の用語は標準化されていないため、地域によって異なる意味になる場合がある。2.6 項参照のこと。
1.1 Background
When a new medicinal product is approved for marketing, demonstration of safety and efficacy are generally based on data from a limited number of patients, many studied under the controlled conditions of randomised trials. Often, higher risk subgroups and patients with concomitant illnesses that require use of other drugs are excluded from clinical trials, and long-term treatment data are limited. Moreover, patients in trials are closely monitored for evidence of adverse events. In clinical practice, monitoring is less intensive, a broader range of patients are treated (age, co-morbidities, drugs, genetic abnormalities), and events too rare to occur in clinical trials may be observed (e.g., severe liver injury). These factors underlie the need for continuing analysis of relevant safety, efficacy,*1 and effectiveness1 information throughout the lifecycle of a medicinal product – promptly, as important findings occur – and periodically – to allow an overall assessment of the accumulating data. Although the majority of new information will be safety-related, new information about effectiveness, limitations of use, alternative treatments, and many other aspects of the drug’s place in therapy may be pertinent to its benefit-risk assessment.
*1 The terms efficacy and effectiveness are not standardised, and have different meanings across some regions. See Section 2.6
ICH E2C ガイドライン「臨床安全性データの取扱い:市販医薬品に関する定期的安全性最新報告」は、1996 年にステップ4 となり、規制当局に対する定期報告の規制要件を調和させることと、共通様式により、承認後一定のタイミングで、医薬品に関する全世界の安全性情報を定期的に報告することを意図していた。当時、定期的安全性最新報告(PSUR)の焦点は、使用患者数を考慮した関連する新規の安全性情報に当てられており、製品の継続的かつ安全な使用方法を最適化するために安全性参照情報*(RSI)に変更が必要かどうかを判断することを目的としていた。
本ガイドラインは、必要な明確化、助言及び柔軟性を示すために2003 年に改訂された。
The ICH Guideline E2C, Clinical Safety Data Management: Periodic Safety Update Reports for Marketed Drugs, achieved Step 4 in 1996, and was intended to harmonise the periodic reporting requirements to regulatory authorities and to provide, in a common format, the worldwide interval safety experience of a medicinal product at defined times post-approval. At that time, the focus of the Periodic Safety Update Report (PSUR) was on relevant new safety information in the context of patient exposure, to determine if changes were needed to the Reference Safety Information* (RSI) in order to optimise the continued safe use of the product. The Guideline was revised in 2003, to provide needed clarification, guidance and flexibility.
それ以降、医薬品安全性監視(pharmacovigilance)を取り巻く環境は進化し、規制当局に提出する多種多様な安全性文書におけるPSUR の役割の再評価が喫緊の課題となった。この再評価により、この分野の進展を考慮しガイドラインをより有用なものとするために、これを改訂しその焦点を再調整することの合意につながるいくつかの要因が明らかとなった。
- 医薬品安全性監視における技術及び科学の著しい進展。例えば、規制当局に対する個別症例安全性報告(ICSR)の電子的提出、自動化されたデータマイニング技術、及びベネフィット・リスク評価に対する関心の高まり
- 能動的かつ文書化されたリスク管理計画のさらなる重視
- 重要な新規のリスク情報の有意義な評価は、医薬品のベネフィットと関連づけて行うべきで あるという認識の高まり
- 医薬品安全性監視に関連する他のICH ガイドラインとの内容の重複
Since that time, the pharmacovigilance environment has evolved, prompting reassessment of the role of the PSUR in the spectrum of safety documents submitted to regulatory authorities. This reassessment highlighted several factors that led to consensus for revision and refocus of the Guideline, to enhance its usefulness in light of advances in the field:
- Significant progress in the technology and science of pharmacovigilance, including electronic submission of Individual Case Safety Reports (ICSRs) to regulatory authorities, automated data mining techniques, and more attention to benefit-risk evaluation;
- Greater emphasis on proactive and documented risk management planning;
- Increasing recognition that meaningful evaluation of important new risk information should be undertaken in the context of a medicinal product’s benefits; and
- Overlap in the content of ICH Guidelines related to pharmacovigilance documentation.
前述のように、PSUR の主たる目的は、既承認医薬品の安全性に関する全体像を示すことにあった。医薬品のリスク評価はベネフィットと関連づけて考えた場合に最も有意義であるとの認識の下に提案されたこの報告書は、特にリスク推定値の重大な変更があった際に、PSUR よりもベネフィットをより重視している。そのような場合、ベネフィット・リスクの全体的かつ系統的な評価が必要となる。したがって、名称は「定期的ベネフィット・リスク評価報告(PBRER)」とされた。PBRER では、新規の情報に注目する一方で、医薬品に関する累積的な知見についても非常に重要視している。
As noted above, the primary objective of the PSUR was to provide a comprehensive picture of the safety of approved medicinal products. With recognition that the assessment of the risk of a medicinal product is most meaningful when considered in light of its benefits, the proposed report would provide greater emphasis on benefit than the PSUR, particularly when risk estimates change importantly. In such cases there will need to be an overall explicit evaluation of benefit-risk. Consequently the name of the proposed report is the “Periodic Benefit-Risk Evaluation Report” (PBRER). The PBRER would also provide greater emphasis on the cumulative knowledge regarding a medicinal product, while retaining a focus on new information.
ベネフィットの正式な評価は、PBRER の新しい特徴の1 つである。しかし、調査期間における安全性又はベネフィット・リスクプロファイルに重要な変更がない限り、通常、ベネフィット評価はその簡潔な考察で十分であると認識されている。そのため、PBRER の特定の項(例:安全性及び有効性に関するデータの評価、安全性シグナル*の評価又はベネフィット・リスク評価)で提供される詳細度は、医薬品の既知もしくは新規で重要なリスクや、新規で重要なベネフィットのエビデンスに対応させる必要がある。
A formal evaluation of benefit is a new feature of the PBRER; however, it is recognised that a concise discussion of benefit will usually be sufficient, unless the safety or benefitrisk profile has changed significantly during the reporting interval. Thus, the level of detail provided in certain sections of the PBRER (e.g., evaluation of safety and efficacy data, evaluation of safety signals,* and benefit-risk evaluation) should be proportional to the medicinal product’s known or emerging important risks and to evidence of emerging important benefits.
PBRER が対象とする範囲は、安全性に加えてベネフィットを含むように拡大されたため、報告に用いる参照情報もこの新たな要素を加味する必要がある。一般に、販売承認取得者(MAH)が以下のような1 つの参照情報源を持つことは非現実的である。
- ベネフィット・リスク評価に供するすべてのパラメータ(すなわち、ベネフィット、有効性/有用性、承認適応及び安全性情報)を含む
- すべてのICH 地域に共通である
- すべての状況に対応している(例:後発医薬品、1 カ国のみで承認されている医薬品)
As the scope of the PBRER has been extended to include benefit as well as safety, the reference information for the report also needs to take this new factor into account. It is generally impractical for Marketing Authorisation Holders (MAHs) to have one reference information source that:
- Encompasses all parameters that contribute towards the benefit-risk evaluation, (i.e., benefit, efficacy/effectiveness, indication(s) and safety information);
- Is common to all ICH regions; and
- Addresses all circumstances, (e.g., generics, products licensed in one country only).
したがって、本ガイドラインでは、PBRER に用いる最も適切な製品参照情報をMAH が選択する際に検討可能な、より現実的な選択肢を提案する。初版のICH E2C の安全性参照情報(例:企業中核安全性情報*[CCSI])の概念に、製品の承認適応の情報を加えるというものである。この製品参照情報は、企業中核データシート(CCDS)*又はMAH が提示するその他の文書である場合がある(2.4 項を参照)。
Therefore, this Guideline proposes more practical options that MAHs can consider in selecting the most appropriate reference product information for the PBRER. These proposals incorporate the original ICH E2C concept of reference safety information (e.g., Company Core Safety Information* [CCSI]), with the addition of the approved indications for the product. This reference product information may be the Company Core Data Sheet* (CCDS) or another document proposed by the MAH (see Section 2.4).
PBRER の17.1 項に要約される調査期間開始時の重要な有効性/有用性情報は、MAH が用いる製品参照情報に関係なく、ベネフィット評価の基準(又は「参照」)となる。
The important baseline efficacy and effectiveness information summarised in Section 17.1 of the PBRER will form the basis (or “reference”) for the benefit evaluation, irrespective of the reference product information used by the MAH.
規制当局への報告の提出頻度は、各国又は地域の規制要件により決定されることから、様々な要因により異なることがある。本ガイドラインには、各地域で様々な頻度で行われるPBRER の提出を管理する際のアドバイスを記載する。
The frequency of submission of reports to regulatory authorities is subject to national or regional regulatory requirements, and may differ, depending on a number of factors. The Guideline includes advice on managing different frequencies of PBRER submission in different regions.
ICH E2C(R1)ガイドライン改訂の背景の1つには、各種規制関連文書の作成に要する作業の重複を減らすことにより効率性を高めるというねらいがあった。このため、本ガイドラインは、PBRER、DSUR(ICH E2F)及びリスク管理計画の安全性検討事項(ICH E2E)の対応する項と同一の内容となるよう作成されている。(1.4 項「PBRER とその他のICH ガイドラインとの関連」を参照のこと。)
One of the motivating factors behind revision of the ICH E2C(R1) Guideline was the desire to enhance efficiency by decreasing the duplication of effort required for the preparation of various regulatory documents. This Guideline has been developed, therefore, such that corresponding sections of the PBRER, Development Safety Update Report (DSUR, ICH E2F), and safety specification of a risk management plan (ICH E2E) can be identical in content. (See also Section 1.4, Relation of the PBRER to Other ICH Documents)
1.2 目的
PBRERの主たる目的は、製品の全体的なベネフィット・リスクプロファイル評価を可能にするため、医薬品のリスク及び承認適応に対するベネフィットに関する新しい情報又は明らかになりつつある情報の包括的、簡潔かつ重要な分析を示すことにある。PBRER には、以下の累積情報に基づき、調査期間にMAH が入手した医薬品に関する新規の情報の評価が含められていなければならない。
- 医薬品のベネフィット・リスクプロファイルに影響を及ぼす可能性がある関連する新たな安全性情報の要約
- 調査期間において入手した重要な新しい有効性/有用性の情報の要約
- 調査期間においてMAH が入手した情報が、医薬品のベネフィット・リスクプロファイルに関する過去の知見に一致しているか否かの検討
- 重要な新しい安全性情報が明らかになりつつある場合には、承認適応に対する総合的なベネフィット・リスク評価の実施
1.2 Objectives
The main objective of a PBRER is to present a comprehensive, concise, and critical analysis of new or emerging information on the risks of the medicinal product, and on its benefit in approved indications, to enable an appraisal of the product’s overall benefitrisk profile. The PBRER should contain an evaluation of new information relevant to the medicinal product that became available to the MAH during the reporting interval, in the context of cumulative information by:
- Summarising relevant new safety information that could have an impact on the benefit-risk profile of the medicinal product;
- Summarising any important new efficacy/effectiveness information that has become available during the reporting interval;
- Examining whether the information obtained by the MAH during the reporting interval is in accord with previous knowledge of the medicinal product’s benefit and risk profile; and
- Where important new safety information has emerged, conducting an integrated benefit-risk evaluation for approved indications.
適切な場合には、PBRER はベネフィット・リスクプロファイルの最適化を目的とした措置の提案を含める。
緊急の安全性情報は、適切な方法により報告する。PBRER が重大な新たな安全性情報の第一報を提供する手段や、新たな安全性の懸念事項*を検出する手段として使われてはならない。
When appropriate, the PBRER should include proposed action(s) to optimise the benefitrisk profile.
Urgent safety information should be reported through the appropriate mechanism; the PBRER is not intended to be used to provide initial notification of significant new safety information or to provide the means by which new safety concerns* are detected.
1.3 PBRER が対象とする範囲
PBRER が主に焦点を当てるのは、国際誕生日*(IBD、世界のいずれかの国で最初の販売承認を取得した日)以降、又は開発国際誕生日(DIBD、いずれの国で介入臨床試験の実施が最初に認可された日)以降に利用可能な情報源から得られた関連する新たな安全性情報3 を該当する有効性/有用性情報の中で評価することである2。調査期間3において確認されたすべての関連する新たな安全性及び有効性/有用性情報は、PBRER の適切な項で考察される。
2 本文書の目的のため、用語「認可」及び「認可された」は臨床試験実施の可否を表し、用語「承認」及び「承認済み」は販売承認を表すものとする。
3 本ガイドラインは、医薬品のベネフィット・リスク評価において提供すべき情報の範囲を制限するものではない。PBRER を提出すべき各国及び地域で適用される法律及び規制を参照のこと。
1.3 Scope of the PBRER
The main focus of each PBRER is the evaluation of relevant new safety information from the available data sources, *2 placed within the context of any pertinent efficacy/effectiveness information that may have become available since the International Birth Date* (IBD), the date of the first marketing approval in any country in the world, or the Development International Birth Date (DIBD), the date of first authorisation for the conduct of an interventional clinical trial in any country. *3 All pertinent new safety and efficacy/effectiveness information discovered during the reporting interval3 should be discussed in the appropriate sections of the PBRER.
*2 For the purpose of this document, the terms “authorisation” and “authorised” refer to clinical trials and the terms “approval” and “approved” refer to marketing applications.
*3 This Guideline should not serve to limit the scope of information to be provided in the evaluation of benefit-risk of a medicinal product. Please refer to the applicable laws and regulations in the countries and regions in which the PBRER is to be submitted.
本ガイドラインの目的において、利用可能な情報源とは、医薬品又はそれに含まれる有効成分に関して、MAH が合理的にアクセスすることが可能な、安全性又はベネフィット・リスクプロファイルの評価に関連する情報のことである(添付資料E「PBRER 作成時に使用する情報源の例」を参照)。例えば、後発医薬品に関しては、MAH が先発医薬品開発会社である製品と比較して得られる情報が少ない可能性があり、また、MAH が依頼者でない臨床試験に関しては公表された報告のみしか入手できない可能性がある。これに対して、MAH が依頼者である臨床試験に関しては、MAH は製品のベネフィット・リスクの評価を目的として患者ごとのデータを入手することが可能であろう。MAH が希望する場合、PBRER の作成に用いる情報源の一覧表をPBRER の添付資料として提供することができる。
For the purposes of this Guideline, sources of available information refer to data regarding the active substance(s) included in the medicinal product or the medicinal product that the MAH may reasonably be expected to have access to, and that are relevant to the evaluation of the safety or benefit-risk profile (see also Appendix E, Examples of Possible Sources of Information that May Be Used in the Preparation of the PBRER). For example, there may be less information available to the MAH regarding a generic product as compared to a product for which the MAH is the innovator, and only a published report may be accessible for a clinical trial not sponsored by the MAH. On the other hand, for a MAH-sponsored clinical trial, the MAH will have access to patient level data towards evaluation of the product’s benefit-risk. When desired by the MAH, a list of the sources of information used to prepare the PBRER can be provided as an appendix to the report.
PBRER は、製品に関する新しい情報に引き続き注目する一方で、累積された知見も記載する。すなわち、全体的な安全性評価及び総合的なベネフィット・リスク評価では累積する情報を考慮する必要がある。医薬品の臨床開発は販売承認後も継続することが多いため、未承認の適応や対象集団に関する市販後の調査又は臨床試験の関連する情報もPBRER に記載する。同様に、医薬品の安全性に関する知見は、適応外使用と関連する情報の評価に由来することがあるため、適切かつ必要な場合には、そのような知見もリスク評価に反映させる。
The PBRER should include cumulative knowledge of the product while retaining focus on new information, i.e., the overall safety evaluation and integrated benefit-risk evaluation will take into account cumulative information. Because clinical development of a drug frequently continues following marketing approval, relevant information from post-marketing studies or clinical trials in unapproved indications or populations should also be included in the PBRER. Similarly, as knowledge of the safety of a medicinal product may be derived from evaluation of data associated with uses other than the approved indication(s), such knowledge would be reflected in the risk evaluation, where relevant and appropriate.
1.4 PBRERとその他のICH ガイドラインとの関連
現在、一部のICH 参加国や参加地域では、各国及び地域の規制要件を満たすことを目的とした各国ごとの定期報告を、承認後のある期間内に提出することを認めている。つまり、既承認の医薬品の安全性に関する定期報告のためのPSUR(ICH E2C ガイドライン(R1))、臨床開発中の医薬品の安全性に関する定期報告DSUR(ICH E2F ガイドライン)並びに承認申請の時点で提出するICH E2E ガイドラ インの安全性検討事項の要素及び/又は医薬品安全性監視活動の計画立案を支援するPSUR の提出である。これらの文書には異なる規制目的、異なる報告頻度があり、単一の規制当局内の異なる部門が文書を審査することもあるため、各文書は単独で完結している必要がある。すなわち、単独で成立する包括的文書であることが必須である。しかし、DSUR、PSUR 及び安全性検討事項の間での内容の重複又は不一致によって、MAH による文書作成に非効率が生じる可能性がある。
1.4 Relation of the PBRER to Other ICH Documents
At present, some ICH countries and regions accept submission of separate types of periodic reports to fulfil national and regional requirements within the post-approval period: the PSUR (ICH Guideline E2C(R1)) for periodic reporting of the safety of approved medicinal products, the DSUR (ICH Guideline E2F) for periodic reporting on the safety of medicinal products that remain in clinical development, and the safety specification component of ICH Guideline E2E that might be submitted at the time of marketing application and/or PSUR submission to aid in the planning of pharmacovigilance activities. As these documents have different regulatory purposes, different periodicities, and can be reviewed by different divisions within a single regulatory authority, each document needs to be complete in its own right – a comprehensive document that can stand alone. Nevertheless, overlap and inconsistencies between the content of the DSUR, PSUR, and safety specification can lead to inefficiencies in the production of the documents by the MAH.
モジュール方式
本ガイドラインでは、個々の項が複数の報告に共通する場合には「モジュール」を使用することにより、異なる規制当局や異なる目的に柔軟に対応することを意図している。すなわち、PBRER はモジュール方式を基本として、いくつかの項の内容がその他の文書の項として使用できるような方法で作成されている。例えば、ICH E2F に提案されているように、ある医薬品のDSUR のDIBD を当該医薬品のPBRER のIBD と一致させた場合、データロックポイント(DLP)が同一であれば、すなわち、各報告書がIBD に基づく1 年間を対象とするとき、DSURの多くの項の内容をPBRER にも用いることが可能である。
Modular Approach
This Guideline aims to facilitate flexibility by encouraging the use of individual sections that are common to more than one report – “modules” that can be used for different regulatory authorities and for different purposes. Therefore, the PBRER has been developed in such a way that the content of several sections may be used for sections of other documents as a basis for a modular approach. For example, if the DIBD of a DSUR for a medicinal product is aligned to the IBD of the PBRER for the same product as suggested in ICH E2F, the content of a number of sections of the DSUR can also be used in the PBRER when the Data Lock Points (DLPs) are the same, i.e., when each report covers an interval of one year based on the IBD.
適切な場合にDSUR(ICH E2F)又はリスク管理計画の安全性検討事項(ICH E2E)のいずれかと共用することができるPBRER の項を本ガイドラインの添付資料D に示した。
モジュール方式としてPBRER、DSUR 及び安全性検討事項の間で共通する項を用いることには以下のような多くの利点がある。
- 複数の規制関連文書間でのモジュールの活用の最大化
- PBRER、DSUR 及び安全性検討事項の間の一貫性の向上
- 作業の不要な重複の回避
- MAH によるこれらの文書の作成における効率の改善
- 例えば、PBRER の対象とする期間が異なる場合や複数の異なる当局に異なる時期に提出する必要がある場合には、既存の項(モジュール)の柔軟な活用を促進する。このような場合、PBRER を提出する際に更新する必要があるのは新しい情報又は新しい評価を含むモジュールのみである。
Appendix D of this Guideline lists the PBRER sections that can be shared with either the DSUR (ICH E2F) or safety specification of a risk management plan (ICH E2E), if appropriate.
The use of common sections across the PBRER, DSUR and safety specification as a modular approach has a number of advantages:
- Maximizes the utility of the modules across multiple regulatory documents;
- Promotes consistency across the PBRER, DSUR and safety specification;
- Avoids unnecessary duplication of effort;
- Is expected to improve efficiency for MAHs in the preparation of these documents;
- Facilitates flexible utilisation of existing sections (modules) when, for example, the PBRER covers different time intervals or needs to be submitted at different times to multiple different authorities. In these circumstances, only modules that include new information or new evaluation would need to be updated when submitting the PBRER.
現時点ではICH E2C(R2)ガイドラインが対象とする範囲ではないが、各種文書間に共通する項について提示されているモジュール方式は、将来、規制当局への提出に用いる電子モジュールの開発を最終的に促すことを期待している。
Although currently out of scope for ICH E2C(R2), it is envisioned that the modular approach proposed, based on common sections across various documents, will ultimately facilitate development of electronic modules for use in future regulatory submissions.
2. 一般原則
2.1 1有効成分に1 つのPBRER
PBRER は、単一のDLP におけるすべての承認適応、剤形及び有効成分に対する投与レジメンに関する情報を記載する。状況によっては、PBRER の項目内において、承認された適応別、剤形別、投与レジメン別又は対象集団別(例:小児対成人)にデータを示すことが適切と考えられる。全く別の承認適応に対して全身投与用及び局所投与用の2 種類の製剤として1 種類の有効成分が使用されるなどの例外的な場合では、PBRER を個別に提出することが適切と考えられる。
これらの場合は、規制当局に必ず相談し、同意を得ること。その時期は、承認時点であることが望ましい。
2. GENERAL PRINCIPLES
2.1 Single PBRER for an Active Substance
The PBRER should provide information on all approved indications, dosage forms, and regimens for the active substance, with a single DLP. In some circumstances, it will be appropriate to present data by indication, dosage form, dosing regimen, or population (e.g., children vs. adults) within the relevant section(s) of the PBRER. In exceptional cases, submission of separate PBRERs might be appropriate, for example, an active substance used in two formulations for systemic and topical administration in entirely different indications. In these cases, the regulatory authorities should be notified and their agreement obtained, preferably at the time of approval.
2.2 配合剤である場合のPBRER
個別に販売されることもある有効成分の組み合わせの場合、配合剤に関する情報は、状況に応じて、単独のPBRER として報告するか、又は各成分のうち1つの成分のPBRER に配合剤の情報として含めて記載することで対応できる。関連するPBRER を一覧として示すことが重要と考えられる。
2.2 PBRERs for Fixed-Dose Combination Product
For combinations of substances also marketed individually, information for the fixed combination may be reported either in a separate PBRER or included as separate presentations in the report for one of the individual substances, depending on the circumstances. Listing related PBRERs is considered important.
2.3 複数の企業が製造及び/又は販売する製品
各MAH は、自社製品のPBRER 提出に対して責任を負う。
企業が契約関係(例:ライセンサーとライセンシーの関係)を締結する場合には、PBRER の作成及び規制当局に対する提出に関して、契約書にそれぞれの責任を明確に定める。
パートナー企業から入手するデータが安全性、ベネフィット及び/又はベネフィット・リスク分析に重要な役割を果たし、報告企業の製品情報に影響を及ぼす場合には、これらのデータをPBRER に記載し、考察する。
2.3 Products Manufactured and/or Marketed by More than One Company
Each MAH is responsible for submitting PBRERs for its own products.
When companies are involved in contractual relationships (e.g., licensor-licensee), respective responsibilities for preparation and submission of the PBRER to the regulatory authorities should be clearly specified in the written agreement.
When data received from a partner company(ies) might contribute meaningfully to the safety, benefit, and/or benefit-risk analyses and influence the reporting company’s product information, these data should be included and discussed in the PBRER.
2.4 参照情報
PBRER の目的は、調査期間において入手した情報が製品のベネフィット及びリスクプロファイルに関する過去の知見と一致しているか否かを評価し、製品参照情報に変更を加えるべきか否かを示すことにある。3 つのICH 地域で利用可能な同一の参照情報があることにより、ベネフィット・リスク評価はより実践的、効率的かつ一貫したものへと促され、PBRER がすべての国や地域で受入れ可能な唯一の報告となるであろう。
2.4 Reference Information
An objective of a PBRER is to evaluate whether information obtained during the reporting interval is in accord with previous knowledge on the product’s benefit and risk profile, and to indicate whether changes should be made to the reference product information. Having one reference source of information that can be applied across the three ICH regions would facilitate a practical, efficient, and consistent approach to the benefit-risk evaluation and make the PBRER a unique report accepted in all countries and regions.
PBRER に用いる製品参照情報は「中核的な安全性情報」及び「承認適応」の要素を含む。PBRER の評価の項における承認適応ごとのベネフィット及びベネフィット・リスク評価を促進するため、製品参照情報の文書にはICH 参加国や参加地域でのすべての承認適応を記載する。これらの承認適応は、ICH 参加国や参加地域以外の国や地域にも該当する可能性が高い。しかし、地域特有の追加の承認適応を有している国にもPBRER を提出しようとする場合、これらの承認適応に関する情報は、MAH が最も適切であると判断した方法で、製品参照情報に追加するか又は地域ごとの添付資料とするかのいずれかで提示する。ベネフィット評価の基準は、PBRER の17.1 項に要約される調査期間開始時の重要な有効性/有用性情報とする。
The reference product information for the PBRER would include “core safety” and “approved indications” components. In order to facilitate the assessment of benefit and benefit-risk by indication in the evaluation sections of the PBRER, the reference product information document should list all approved indications in ICH countries or regions. It is likely that these indications will also apply in other countries or regions. However, when the PBRER is also to be submitted to other countries in which there are additional locally approved indications, these indications may either be added to the reference product information or handled as a regional appendix/appendices as considered most appropriate by the MAH. The basis for the benefit evaluation should be the baseline important efficacy/effectiveness information summarised in Section 17.1 of the PBRER.
PBRER に用いる製品参照情報は「中核的な安全性情報」及び「承認適応」の要素を含む。PBRER の評価の項における承認適応ごとのベネフィット及びベネフィット・リスク評価を促進するため、製品参照情報の文書にはICH 参加国や参加地域でのすべての承認適応を記載する。これらの承認適応は、ICH 参加国や参加地域以外の国や地域にも該当する可能性が高い。しかし、地域特有の追加の承認適応を有している国にもPBRER を提出しようとする場合、これらの承認適応に関する情報は、MAH が最も適切であると判断した方法で、製品参照情報に追加するか又は地域ごとの添付資料とするかのいずれかで提示する。ベネフィット評価の基準は、PBRER の17.1 項に要約される調査期間開始時の重要な有効性/有用性情報とする。
The reference product information for the PBRER would include “core safety” and “approved indications” components. In order to facilitate the assessment of benefit and benefit-risk by indication in the evaluation sections of the PBRER, the reference product information document should list all approved indications in ICH countries or regions. It is likely that these indications will also apply in other countries or regions. However, when the PBRER is also to be submitted to other countries in which there are additional locally approved indications, these indications may either be added to the reference product information or handled as a regional appendix/appendices as considered most appropriate by the MAH. The basis for the benefit evaluation should be the baseline important efficacy/effectiveness information summarised in Section 17.1 of the PBRER.
PBRER に用いる最も適切な製品参照情報を選択する際、MAH は以下の選択肢を検討することができる。
- 企業中核データシート
ICH E2C(R1)の提案事項に従い、MAH が独自のCCDS を作成することは一般的に行われている。CCDS は、安全性、承認適応、用法・用量、薬理その他医薬品に係る情報に関する項を含む。CCDS に含まれる中核的な安全性情報は、CCSI と呼ばれる。PBRER のリスクに関する項及びベネフィットが評価される主な承認適応の両方について、調査期間末の時点で有効な最新のCCDS を製品参照情報として用いることがMAH にとって現実的な選択肢である。
医薬品のCCDS に承認適応に関する情報が含まれない場合、MAH はPBRER における承認適応のための参照情報として用いた文書を明記する。
- 製品参照情報に関する他の選択肢
製品にCCDS 又はCCSI が存在しない場合、例えば、1カ国若しくは1 地域のみで承認されている製品又は長年販売されている既存品や後発品の場合、MAH は、使用している参照情報を明記する。このような情報には、米国の添付文書(USPI)、欧州の製品概要(SmPC)又は日本の添付文書などの各国又は地域の製品情報が必要に応じて含まれる。ベネフィット評価の基準は、PBRER の17.1 項に要約される調査期間開始時の重要な有効性/有用性情報とする。
承認適応に関する参照情報がRSI とは異なる文書である場合、PBRER のDLP 時点での最新版の当該参照情報を添付資料1 に記載する。
MAH は、調査期間中に新しい安全性情報を入手したときは随時、製品参照情報又はRSI の改訂が必要か否かを継続的に評価する。調査期間中に行った製品参照情報又はRSI の重要な改訂は、PBRER の4 項「安全性参照情報の変更」に記載する。例えば、以下の点が含まれる。
- RSI の禁忌、警告/注意の項の変更
- 副作用又は相互作用の追加
- 過量投与に関する重要な新しい情報の追加
- 安全性上の問題又は有効性の欠如を理由とする承認適応の取り消し又はその他の制限
DLP 後であるがPBRER の提出前に行われたRSI の重要な変更については、可能であればPBRER の14 項「データロックポイント後に入手した情報」に記載する。
該当する地域の規制要件に規定されている場合、MAH は地域毎の添付資料に、各国又は地域の承認済み製品情報への最終的な変更、継続中の変更又は予定する変更に関する情報も記載する。
The following possible options can be considered by MAHs in selecting the most appropriate reference product information for a PBRER:
- Company Core Data Sheet
In accordance with ICH E2C(R1) recommendations, it is a common practice for MAHs to prepare their own CCDS, which includes sections relating to safety, indications, dosing, pharmacology, and other information concerning the medicinal product. The core safety information contained within the CCDS is referred to as the CCSI. A practical option is for MAHs to use the latest CCDS in effect at the end of the reporting interval as the reference product information for both the risk sections of the PBRER as well as the main approved indications for which benefit is evaluated.
When the CCDS for a medicinal product does not contain information on approved indications, the MAH should clearly specify which document is used as the reference information for the approved indications in the PBRER.
- Other options for the reference product information
When there is no CCDS or CCSI for a product, e.g., where the product is approved in only one country or region, or for established/generic products on the market for many years, the MAH should clearly specify the reference information being used. This may comprise national or regional product information such as the US Package Insert (USPI) or European Summary of Product Characteristics (SmPC), or the Japanese package insert, as appropriate. The basis for the benefit evaluation should be the baseline important efficacy/effectiveness information summarised in Section 17.1 of the PBRER.
Where the reference information for approved indications is a separate document to the RSI, the version current at the DLP of the PBRER should be included in Appendix 1.
The MAH should continuously evaluate whether any revision of the reference product information/RSI is needed whenever new safety information is obtained throughout the reporting interval. Significant changes to the reference product information/RSI made during the interval should be described in Section 4 of the PBRER (“Changes to Reference Safety Information”) and include:
- Changes to contraindications, warnings/precautions sections of the RSI;
- Addition of Adverse Drug Reaction(s) (ADR) and interactions;
- Addition of important new information on use in overdose; and
- Removal of an indication or other restrictions for safety or lack of efficacy reasons.
Significant changes to the RSI made after the DLP but before submission of the PBRER should be included in Section 14 of the report (Late-Breaking Information), if feasible.
If stipulated by applicable regional requirements, the MAH should provide, in a regional appendix, information on any final, ongoing, or proposed changes to the national or local authorised product information.
2.5 PBRER 内における詳細度
PBRER の特定の項目における詳細度は、医薬品の既知又は明確になりつつある重要なベネフィット又はリスクに依存する。この考え方は、安全性データ、有効性/有用性データ、安全性シグナル及びベネフィット・リスクの評価が行われるPBRER の項にも当てはまる。したがって、このようなPBRER の各項で示される情報の詳細度は、個々のPBRER 間で異なる。
2.5 Level of Detail Within PBRER
The level of detail provided in certain sections of the PBRER should depend on the medicinal product’s known or emerging important benefits and risks. This approach is applicable to those sections of the PBRER in which there is evaluation of safety data, efficacy/effectiveness data, safety signals, and benefit-risk. Therefore, the extent of information provided in such PBRER sections will vary among individual PBRERs.
例えば、重要な新しい安全性情報が存在する場合、頑健なベネフィット・リスク分析を促進するためには、そのような情報の詳細な記載に加え、関連するベネフィットの情報の記載が必要である。反対に、調査期間において重要な新しい安全性情報がほとんど入手できなかった場合には、ベースライン時のベネフィット情報の簡潔な要約で十分であり、ベネフィット・リスク評価は主として、直近の調査期間の安全性データの評価から構成される。
For example, when there is important new safety information, a detailed presentation of that information should be included, plus the relevant benefit information, in order to facilitate a robust benefit-risk analysis. Conversely, when little new important safety information has become available during the reporting interval, a concise summary of baseline benefit information should be sufficient, and the benefit-risk evaluation would consist primarily of an evaluation of updated interval safety data.
2.6 有効性/有用性
本文書の目的のため、臨床試験及び日常診療におけるベネフィットに関するエビデンスを報告する。「有効性/有用性」の用語が地域間で統一されていないため、本ガイドラインでは、臨床試験及び日常診療の両方から得られた情報は、PBRER に含めるべきベネフィットに関する情報の範囲内であることを明確にするために「有効性/有用性」の用語を用いる。地域によっては、有効性は比較臨床試験からのベネフィットに関するエビデンスを指し、有用性は日常診療での使用を意味するが、この区別がない地域もある。
2.6 Efficacy/Effectiveness
For the purpose of this Guideline, evidence on benefits in clinical trials and in everyday medical practice should be reported. Because the terms are not harmonized across regions, the terms “efficacy/effectiveness” are used in this Guideline to clarify that information from both clinical trials and everyday medical practice are within the scope of the information on benefit to be included within the PBRER. In some regions, efficacy refers to evidence of benefit from controlled clinical trials while effectiveness implies use in everyday medical practice. Conversely, in other regions, this distinction is not made.
2.7 ベネフィット・リスク評価
医薬品が販売承認された際には、承認された製品情報に準拠して使用する場合には医薬品のベネフィットがそのリスクを上回るという結論に達したことを意味する。市販後に医薬品に関する新しい情報が明らかとなるに伴い、ベネフィットが引き続きリスクを上回るか否かを判断し、リスク最小化活動(例:製品情報の変更、処方者への情報伝達又はその他の措置)を通じてベネフィット・リスクのバランスを改善する措置が必要か否かを検討するため、ベネフィット・リスク評価を実施する必要がある。
2.7 Benefit-Risk Evaluation
When a drug is approved for marketing, a conclusion has been reached that, when used in accordance with approved product information, its benefits outweigh its risks. As new information about the drug emerges during marketing experience, benefit-risk evaluation should be carried out to determine whether benefits continue to outweigh risks, and to consider whether steps need to be taken to improve the benefit-risk balance through risk minimisation activities, e.g., labelling changes, communications with prescribers, or other steps.
2.8 報告頻度とPBRER のデータロックポイント
2.8.1 国際誕生日とデータロックポイント
各医薬品には1 つのIBD を用いることとする。IBD とは世界のいずれかの国で有効成分を含む製剤に最初の販売承認が会社に与えられた日付である。報告が、様々な剤形、処方又は使用方法(承認適応、投与経路及び/又は対象集団)に関する情報を含む場合には、各種承認のうち最初に販売承認を取得した日をIBD とみなし、PBRER 作成の目的のためにDLP を定めることとする。DLP は、PBRER に記載されるべきデータのカットオフとして指定された日付である。共通のIBD に基づき調和させたDLP を用いてPBRER を作成することにより、全世界のすべての規制当局が同一かつ最新の安全性及びベネフィット・リスク情報を審査することが可能になる。
2.8 Periodicity and PBRER Data Lock Point
2.8.1 International Birth Date and Data Lock Point
Each medicinal product should have an IBD: the IBD is the date of the first marketing approval for any product containing the active substance granted to any company in any country in the world. When a report contains information on different dosage forms, formulations, or uses (indications, routes and/or populations), the date of the first marketing approval for any of the various authorisations should be regarded as the IBD and, therefore, determine the DLP for purposes of the PBRER. The DLP is the date designated as the cut-off for data to be included in a PBRER. Through PBRERs prepared with harmonised DLPs based on a common IBD, the same updated safety and benefit-risk information can be reviewed globally by different regulatory authorities.
1つの配合剤に対し単独のPBRER を作成する場合(2.2 項参照)、当該PBRER のDLP は、含まれる有効成分のうちの1 つの最も早いIBD、又は当該配合剤が世界のいずれかの場所で最初の販売承認を取得した日であるIBD のいずれに基づくことも可能である。
医薬品の臨床開発が販売承認後も継続する場合、依頼者/MAH が希望する場合は、DSUR とPBRER の両方を同時に作成できるように、同一のDLP を用いてDSUR の調査期間の開始時点をIBD に基づくサイクルと同期させることができる。この方法を用いることにより、PBRER とDSUR を毎年提出する際に、提示する共通の項又はモジュールをPBRER とDSUR の両方に用いることが促進される(添付資料D 参照)。
When a separate PBRER is prepared for a fixed-dose combination product (see Section 2.2), the DLP for that PBRER can be based on either the earliest IBD of one of the component active substances, or the IBD of the first marketing approval anywhere in the world for the fixed-dose combination.
When clinical development of a medicinal product continues following marketing approval, if desired by the sponsor/MAH, the beginning of the DSUR reporting interval can be synchronized with the IBD-based cycle, so that both the DSUR and PBRER can be prepared at the same time, using the same DLP. This approach will facilitate use of the proposed common sections/modules for both the PBRER and DSUR when both are submitted annually (see Appendix D).
2.8.2 提出頻度の異なるPBRER の扱い
PBRER の提出の必要性と規制当局に対する報告提出の頻度は、各国又は地域の規制要件に従うとともに、通常、承認日や製品が販売されている期間、製品のベネフィット・リスクプロファイルに関する知見の程度などの要因に依存する。PBRER の様式及び内容は、6 カ月以上の報告を対象とする定期報告に適用することを意図している。数年にわたり医薬品が販売されていると、各国又は地域の規制は、間隔を延長した提出頻度の設定を認めることがある。例えば、確立し、かつ許容可能なプロファイルを示すと判断された製品又はリスクが低いと判断された製品の場合には、1 年を超える間隔での提出を認めることがある。しかし、地域によってはより頻繁なPBRER 提出を引き続き求めることもある。結果として、MAH にとっては以下の状況が考えられる。
- 異なる地域から同時に6 カ月ごと、1 年ごと又はより低頻度の提出スケジュールでPBRER の提出を求められることがある。
- 臨床的な使用方法の重要な追加又は変更が承認された後(例:新しい承認適応及び/又は新しい投与対象集団の追加)に報告頻度の変更が適用されることもある。このような状況では、PBRER 提出頻度が過去に減じられた古い製品であっても、調査期間が短縮されることがある。
- 規制当局によって臨時のPBRER が求められることがある(本ガイドラインの8.2.1 項を参照)。
2.8.2 Managing Different Frequencies of PBRER Submission
The need for the submission of a PBRER and the frequency of report submission to regulatory authorities are subject to national or regional regulatory requirements, and usually depend on such factors as approval dates, the length of time the product has been on the market, and the extent of knowledge of the benefit-risk profile of the product.
The PBRER format and content are intended to apply to periodic reports that cover reporting periods of 6 months or longer. Once a drug has been marketed for several years, national or regional regulation may allow the frequency of submission to be extended to longer time intervals, e.g., greater than one year for products considered to have an established and acceptable profile or considered to be low risk; however, more frequent PBRERs may continue to be required in other regions. As a result, the following scenarios may be encountered by MAHs:
- PBRERs may be required on 6-monthly, annual, and less frequent submission timetables simultaneously across different regions.
- Changes in reporting frequency may also apply after important additions or changes in clinical use are approved (e.g., new indication[s] and/or new population[s]). In these circumstances, it is possible that the reporting interval will be shortened, even for older products with a previously reduced frequency of PBRER submission.
- An ad hoc PBRER may be requested by a regulatory authority (see Section 2.8.2.1 of this Guideline).
報告が対象とする調査期間の長さにかかわらず、
- 各PBRER は単独で成立する文書であり、MAH が現在入手している新規の及び累積的な情報を反映するものである。
- 規制当局は、通常、PBRER のDLP を定めるために、IBD を用いることを認める。各国又は地域の要件がこれと異なる場合、MAH は当該規制当局に相談することができる。各製品に対して調和した単一のIBD 及びDLP を使用することは、PBRER 作成に伴う作業負荷を軽減するうえで重要であり、PBRER の本来の目的、すなわち、異なる規制当局に提出できる1 製品についての世界で唯一の概要を作成することを尊重している。
- 新たに承認された製品の場合、承認後、少なくとも最初の2 年間にわたり、6 カ月ごとの報告が多くの地域で適用される。
- 定期的に提出されるPBRER の場合、報告は、6 カ月の調査期間のデータセット又はその倍数の累積データに基づく必要がある。
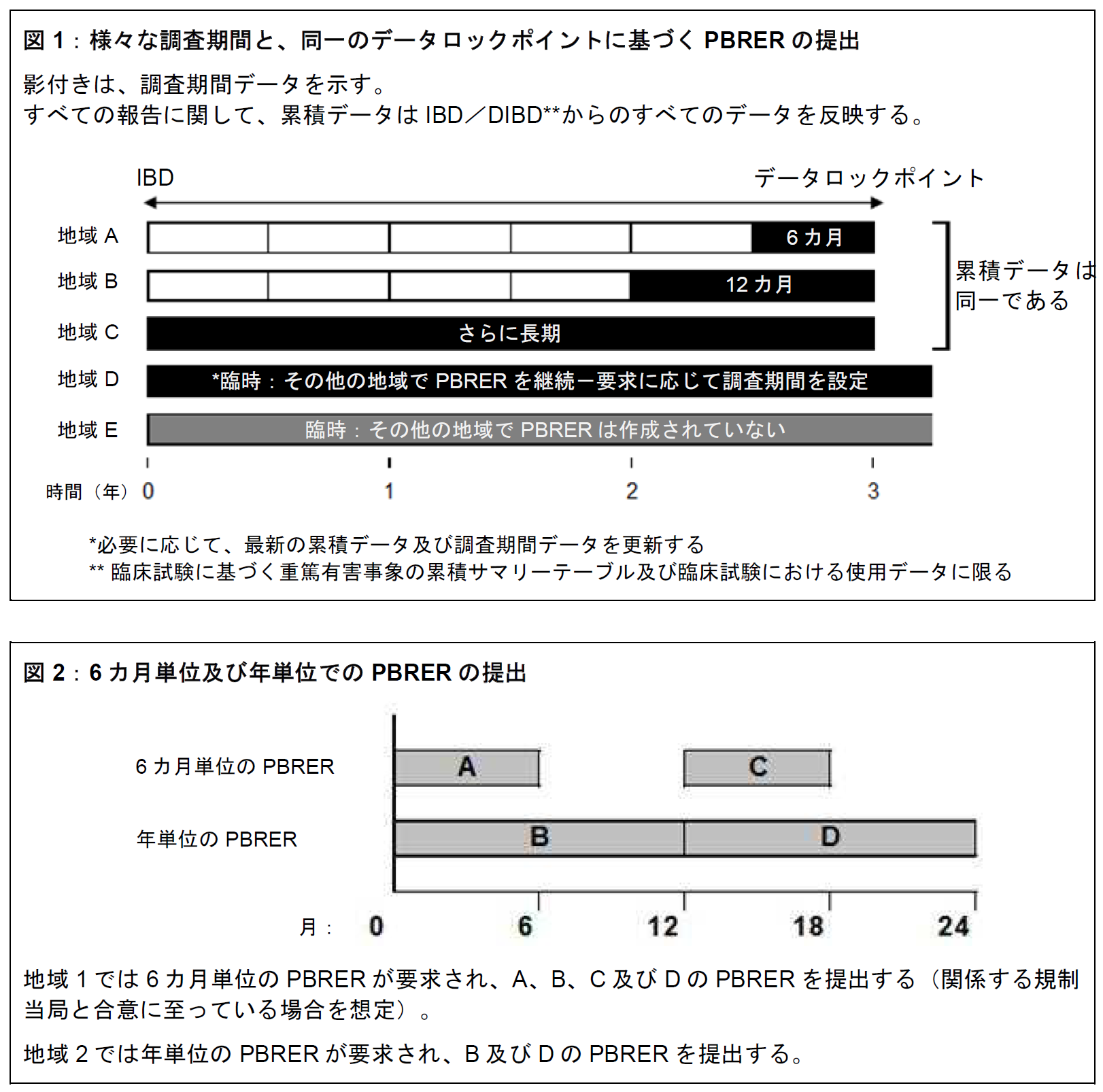
- 各PBRER の調査期間情報を提示する項は、更新する必要性が生じやすいが、前回のPBRER 作成以降に新しい情報が発生していない項については、適切な場合、前回のPBRER で使用した内容をレビューし、これを再利用することができる。レビューを実施し、内容が現在の情報に合った最新の状態であれば、累積データの評価を提示する項を更新する必要はないと判断することができる。図1 を参照のこと。
- MAH が異なる規制当局に対して6 カ月及び年単位の両方でPBRER を作成している場合、6 カ月ごとのPBRER を求める規制当局に対して12 カ月間のデータを含むPBRER を提出することが認められる場合がある。図2 を参照のこと。MAH は、この方法が受け入れられるかについて関係する規制当局と相談する。
Independent of the length of the interval covered by the report:
- Each PBRER should be stand-alone and reflect new and cumulative information currently available to the MAH.
- Regulators will normally accept use of the IBD to determine the DLP for PBRERs. Where national or regional requirements differ from this, the MAH may wish to discuss with the relevant regulatory authority. Use of a single harmonised IBD and DLP for each product is important in order to reduce the burden of work involved in preparing PBRERs, and respects the original purpose of the PBRER – to prepare a single worldwide summary on a product that can be submitted to different regulatory authorities.
- For newly approved products, a 6-monthly periodicity applies in many regions, for at least the first 2 years after approval.
- For PBRERs submitted on a routine/regular basis, the reports should be based on cumulative data, with interval data sets of 6 months, or multiples thereof.
- Sections that provide interval information are likely to need to be updated for each PBRER, and the content used in the previous PBRER can be reviewed and reused for sections where no new information has arisen since preparation of the last PBRER, if appropriate. Following review, it may be determined that sections providing evaluation of cumulative data may not need to be updated if the content remains up to date with current information. See Figure 1.
- In situations when an MAH is preparing PBRERs on both a six-monthly and annual basis for different regulatory authorities, the regulatory authority requiring a PBRER on a six-month cycle may accept PBRERs containing 12-month interval data. See Figure 2. MAHs should discuss the acceptability of this approach with the relevant regulatory authority(ies).






