


ザイリックの販売、導入支援、ユーザーサポート
ZYLiQ Sales, Installation Support, and User Support
![]()
ザイリック (ZYLiQ) とは
ZYLiQは、米国Symbiance社が開発したアプリケーションです。ZYLiQは、人工知能(AI)を利用して、治験総括報告書(CSR)や治験に関わる科学的文書の作成を自動化します。
治験総括報告書(CSR)は、治験の方法と結果について説明する、メディカルライターにより執筆される長大な文書(通常、本文は100~300ページ、付録部分は数百~数千ページ)です。通常、この文書の執筆には多くの時間がメディカルライターの手作業で費やされます。メディカルライターは、治験実施計画書(Protocol)、統計解析計画書(SAP)、症例報告書(CRF)、SAE報告書などのソース文書から相当量の情報を取り込みながらCSRを作成します。
ZYLiQは、治験実施計画書、SAP、SAE報告書などのソース文書から、ICH E3のCSRガイドラインに従って適切なセクションの情報を自動的に抽出し、照合を行います。ZYLiQは、自動化によりメディカルライターの多大な労力と時間を軽減します。また、ZYLiQにはワークフローとダッシュボードモジュールがあり、スポンサーのニーズに応じて設定することができます。
メリット
- 本システムは、プロトコール、SAP、その他のソースからの情報を用いて、ICH-E3ガイドラインに基づいたCSRを生成するように設計されており、メディカルライターの作業時間を60%~70%節約することができます。
- メディカルライターは、試験結果の解釈とその論点に集中することができます。
- ワークフロー統合では、スポンサーのニーズに応じてシステムを構成することができます。
ZYLiQの販売代理店
当社は、米国のSymbiance社のアプリケーションソフトウェアZYLiQの日本での販売代理店として、ZYLiQのプレゼンテーション、受注、納品、導入支援、ユーザーサポート業務を日本語で行っております。当社では、請求書、納品書などの各書類を、お客様の指定フォーマットで発行することも可能です。


About ZYLiQ
ZYLiQ is an application developed by Symbiance Inc to automate the writing of CSR and Scientific documents created by Medical Writers, with the help of Artificial Intelligence (AI).
Clinical Study Report (CSR) is a lengthy document (typically 100-300 pages for the main text and hundreds to thousands of pages for the appendices) written by a medical writer that describes the methods and results of the clinical trial. Authoring this document consumes more manual effort. The source documents such as Protocol, Statistical Analysis Plan (SAP), Case Report Form (CRF), Safety Narratives, In-text tables, Tables, Listings and Figures (TLFs) contribute considerable amount of information to the CSR.
The tool automatically extracts and collates information from source documents such as protocol, SAP, Safety Narratives etc., in the appropriate sections as per ICH E3 guidelines for CSR. The tool automates significant effort of the medical writers. The tool also has the workflow & dashboard module which can be configured as per the sponsor needs.
Benefits
- The system is designed to generate pre-filled CSR with information from Protocol, SAP and other sources as per ICH-E3 guidelines in the respective and system well interpret some of the study result sections which could save medical writers 60%-70% of time.
- Medical writers can focus more on interpretation of study results and their discussion points.
- In workflow integration, system can be configured as per the sponsor’s needs.
ZYLiQ Sales Agent in Japan
As a distributor in Japan for ZYLiQ application software from Symbiance Inc. of the U.S., MedicaLingual provides ZYLiQ presentation, order receipt, delivery, installation support, and user support services in Japanese language. MedicaLingual can also issue invoices, delivery slips, and other documents in the format specified by the customer.
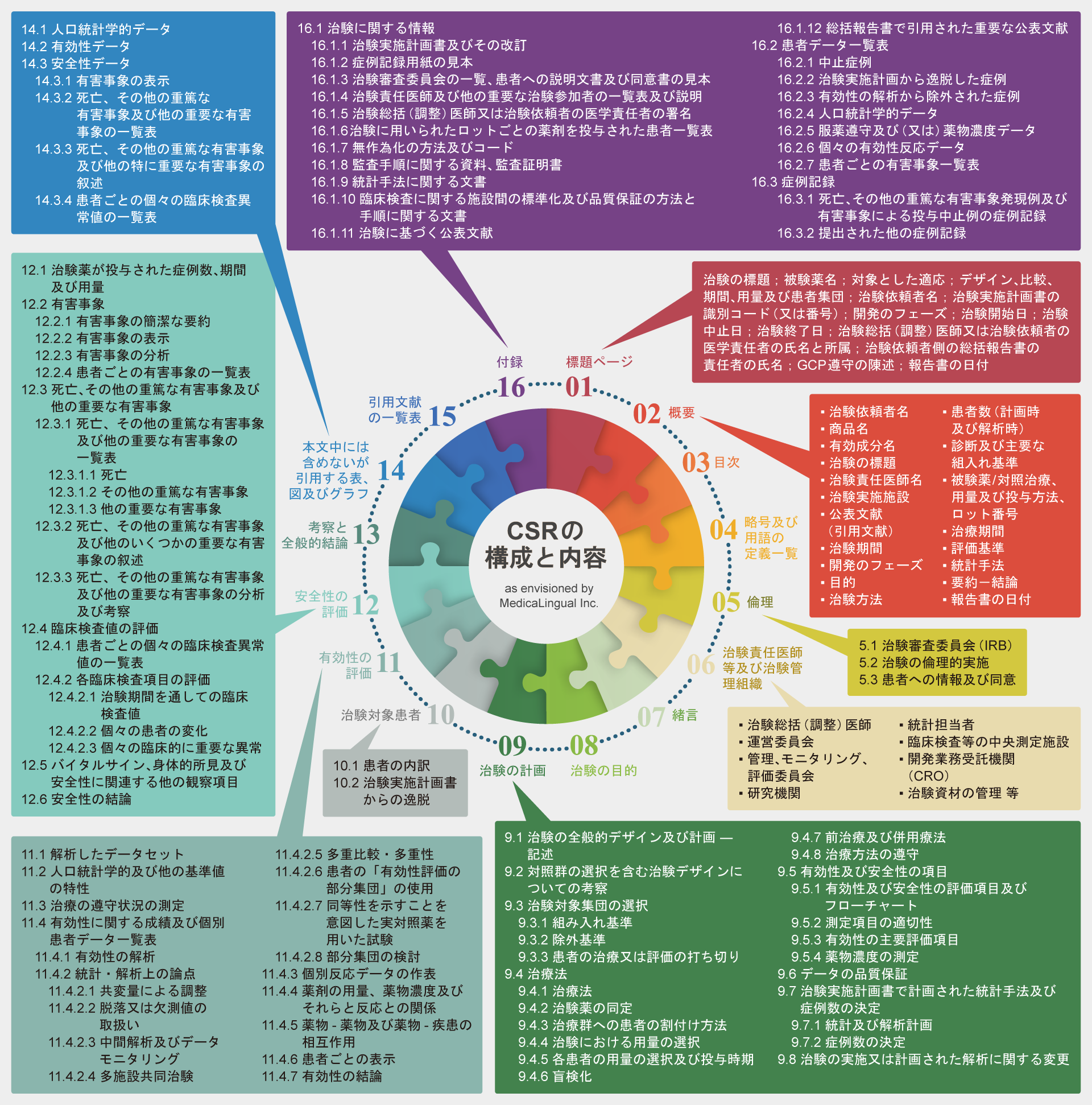
治験の総括報告書の構成と内容(Structure & Content)― 薬審第335号
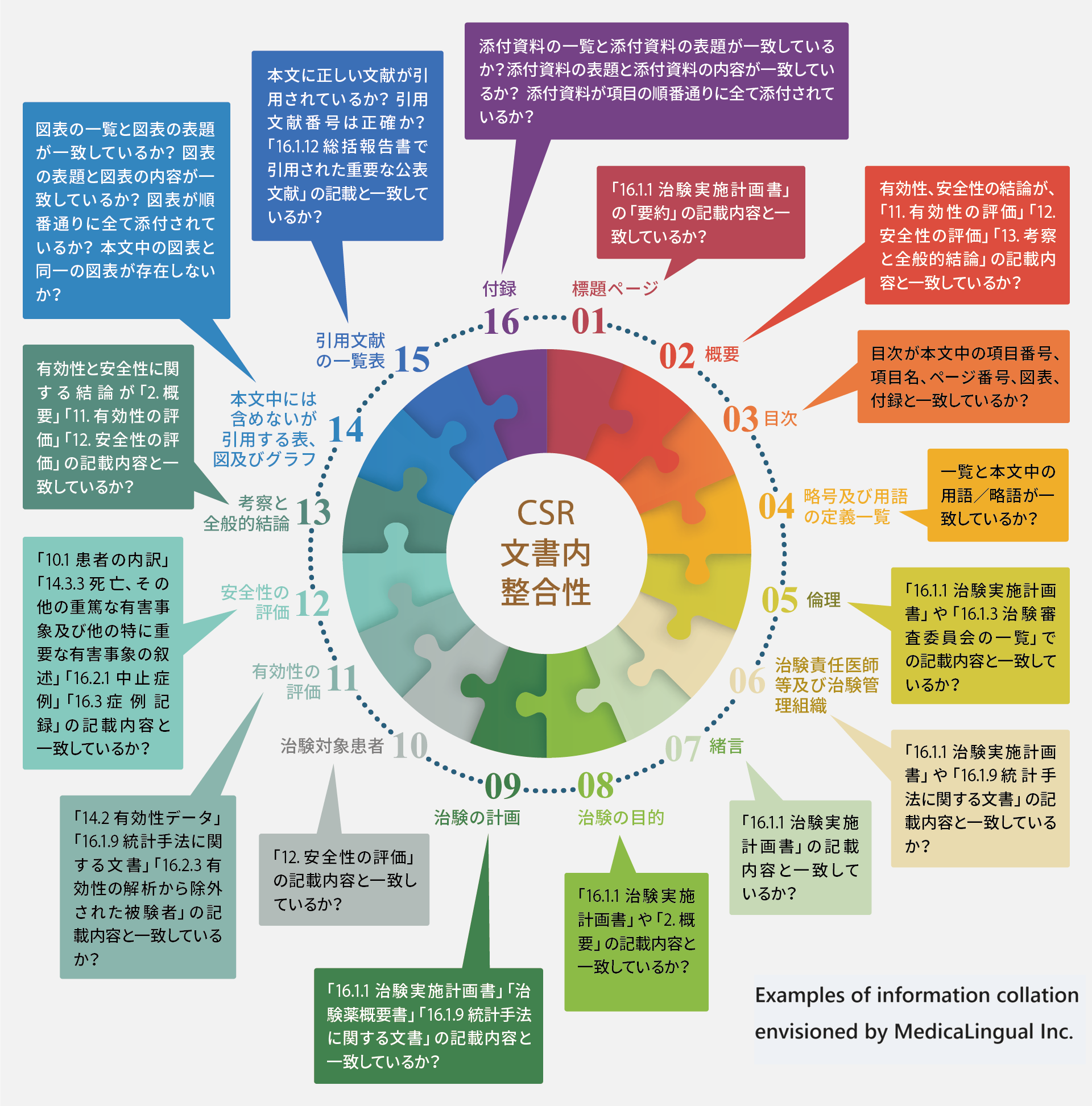
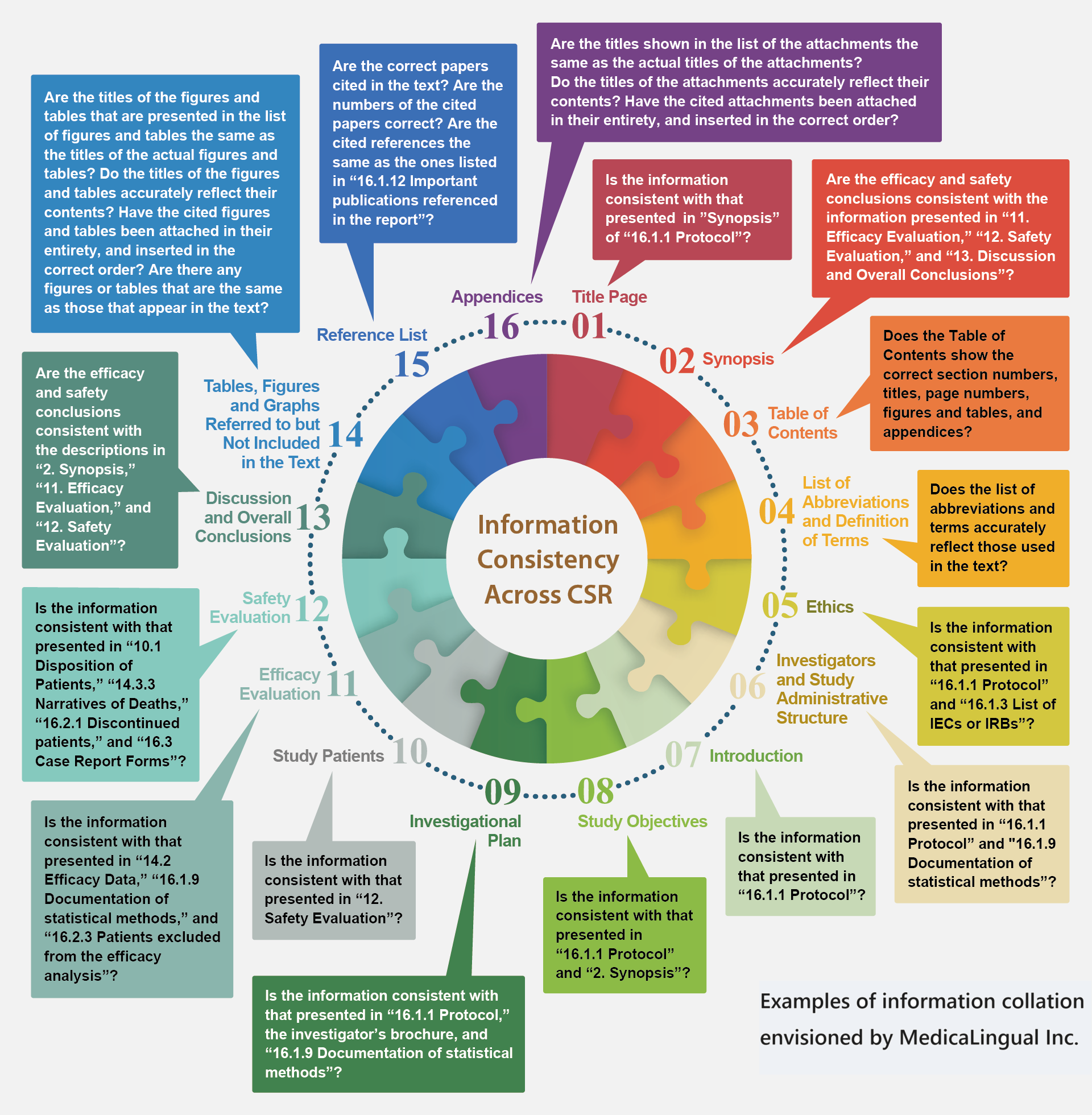
CSRの文書内の整合性
ZYLiQは、ソース文書から、ICH E3のCSRガイドラインに従って、適切なセクションの情報を自動的に抽出し照合を行います。
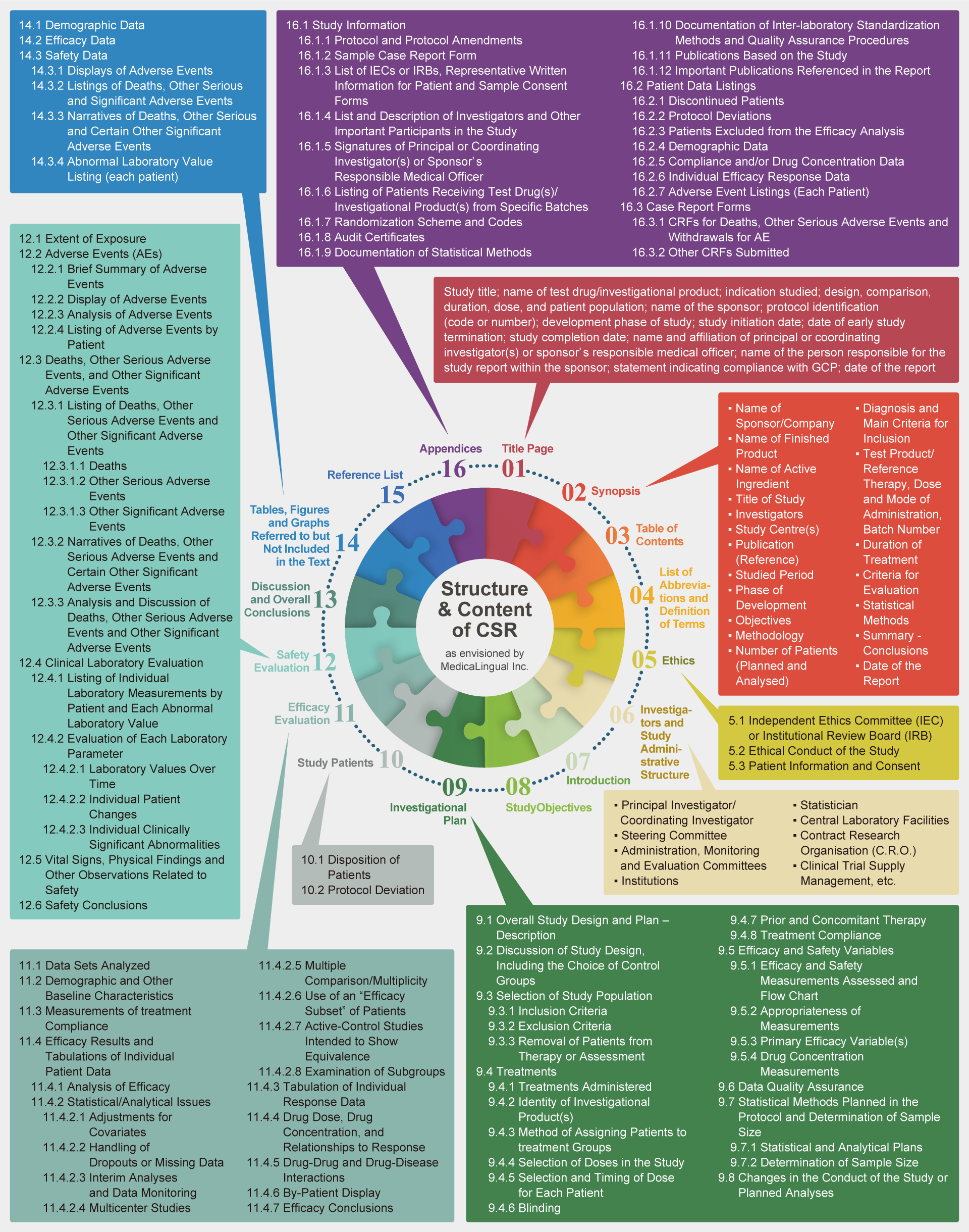
Structure and Content of Clinical Study Reports ― ICH-E3
Information Consistency Across CSR
ZYLiQ automatically extracts and collates information from source documents in the appropriate sections as per ICH E3 guidelines for CSR.
薬審第335号:治験の総括報告書の構成と内容
ICH-E3: Structure and Content of Clinical Study Reports
| 1 | 標題ページ | 1 | Title Page |
| 2 | 概要 | 2 | Synopsis |
| 3 | 目次 | 3 | Table of Contents for the Individual Clinical Study Report |
| 4 | 略号及び用語の定義一覧 | 4 | List of Abbreviations and Definition of Terms |
| 5 | 倫理 | 5 | Ethics |
| 5.1 | 治験審査委員会(IRB) | 5.1 | Independent Ethics Committee (IEC) or Institutional Review Board (IRB) |
| 5.2 | 治験の倫理的実施 | 5.2 | Ethical Conduct of the Study |
| 5.3 | 患者への情報及び同意 | 5.3 | Patient Information and Consent |
| 6 | 治験責任医師等及び治験管理組織 | 6 | Investigators and Study Administrative Structure |
| 7 | 緒言 | 7 | Introduction |
| 8 | 治験の目的 | 8 | Study Objectives |
| 9 | 治験の計画 | 9 | Investigational Plan |
| 9.1 | 治験の全般的デザイン及び計画―記述 | 9.1 | Overall Study Design and Plan – Description |
| 9.2 | 対照群の選択を含む治験デザインについての考察 | 9.2 | Discussion of Study Design, Including the Choice of Control Groups |
| 9.3 | 治験対象集団の選択 | 9.3 | Selection of Study Population |
| 9.3.1 | 組み入れ基準 | 9.3.1 | Inclusion Criteria |
| 9.3.2 | 除外基準 | 9.3.2 | Exclusion Criteria |
| 9.3.3 | 患者の治療又は評価の打ち切り | 9.3.3 | Removal of Patients from Therapy or Assessment |
| 9.4 | 治療法 | 9.4 | Treatments |
| 9.4.1 | 治療法 | 9.4.1 | Treatments Administered |
| 9.4.2 | 治験薬の同定 | 9.4.2 | Identity of Investigational Product(s) |
| 9.4.3 | 治療群への患者の割付け方法 | 9.4.3 | Method of Assigning Patients to treatment Groups |
| 9.4.4 | 治験における用量の選択 | 9.4.4 | Selection of Doses in the Study |
| 9.4.5 | 各患者の用量の選択及び投与時期 | 9.4.5 | Selection and Timing of Dose for Each Patient |
| 9.4.6 | 盲検化 | 9.4.6 | Blinding |
| 9.4.7 | 前治療及び併用療法 | 9.4.7 | Prior and Concomitant Therapy |
| 9.4.8 | 治療方法の遵守 | 9.4.8 | Treatment Compliance |
| 9.5 | 有効性及び安全性の項目 | 9.5 | Efficacy and Safety Variables |
| 9.5.1 | 有効性及び安全性の評価項目及びフローチャート | 9.5.1 | Efficacy and Safety Measurements Assessed and Flow Chart |
| 9.5.2 | 測定項目の適切性 | 9.5.2 | Appropriateness of Measurements |
| 9.5.3 | 有効性の主要評価項目 | 9.5.3 | Primary Efficacy Variable(s) |
| 9.5.4 | 薬物濃度の測定 | 9.5.4 | Drug Concentration Measurements |
| 9.6 | データの品質保証 | 9.6 | Data Quality Assurance |
| 9.7 | 治験実施計画書で計画された統計手法及び症例数の決定 | 9.7 | Statistical Methods Planned in the Protocol and Determination of Sample Size |
| 9.7.1 | 統計及び解析計画 | 9.7.1 | Statistical and Analytical Plans |
| 9.7.2 | 症例数の決定 | 9.7.2 | Determination of Sample Size |
| 9.8 | 治験の実施又は計画された解析に関する変更 | 9.8 | Changes in the Conduct of the Study or Planned Analyses |
| 10 | 治験対象患者 | 10 | Study Patients |
| 10.1 | 患者の内訳 | 10.1 | Disposition of Patients |
| 10.2 | 治験実施計画書からの逸脱 | 10.2 | Protocol Deviation |
| 11 | 有効性の評価 | 11 | Efficacy Evaluation |
| 11.1 | 解析したデータセット | 11.1 | Data Sets Analyzed |
| 11.2 | 人口統計学的及び他の基準値の特性 | 11.2 | Demographic and Other Baseline Characteristics |
| 11.3 | 治療の遵守状況の測定 | 11.3 | Measurements of treatment Compliance |
| 11.4 | 有効性に関する成績及び個別患者データ一覧表 | 11.4 | Efficacy Results and Tabulations of Individual Patient Data |
| 11.4.1 | 有効性の解析 | 11.4.1 | Analysis of Efficacy |
| 11.4.2 | 統計・解析上の論点 | 11.4.2 | Statistical/Analytical Issues |
| 11.4.2.1 | 共変量による調整 | 11.4.2.1 | Adjustments for Covariates |
| 11.4.2.2 | 脱落又は欠測値の取扱い | 11.4.2.2 | Handling of Dropouts or Missing Data |
| 11.4.2.3 | 中間解析及びデータモニタリング | 11.4.2.3 | Interim Analyses and Data Monitoring |
| 11.4.2.4 | 多施設共同治験 | 11.4.2.4 | Multicenter Studies |
| 11.4.2.5 | 多重比較・多重性 | 11.4.2.5 | Multiple Comparison/Multiplicity |
| 11.4.2.6 | 患者の「有効性評価の部分集団」の使用 | 11.4.2.6 | Use of an “Efficacy Subset” of Patients |
| 11.4.2.7 | 同等性を示すことを意図した実対照薬を用いた試験 | 11.4.2.7 | Active-Control Studies Intended to Show Equivalence |
| 11.4.2.8 | 部分集団の検討 | 11.4.2.8 | Examination of Subgroups |
| 11.4.3 | 個別反応データの作表 | 11.4.3 | Tabulation of Individual Response Data |
| 11.4.4 | 薬剤の用量、薬物濃度及びそれらと反応との関係 | 11.4.4 | Drug Dose, Drug Concentration, and Relationships to Response |
| 11.4.5 | 薬物―薬物及び薬物―疾患の相互作用 | 11.4.5 | Drug-Drug and Drug-Disease Interactions |
| 11.4.6 | 患者ごとの表示 | 11.4.6 | By-Patient Display |
| 11.4.7 | 有効性の結論 | 11.4.7 | Efficacy Conclusions |
| 12 | 安全性の評価 | 12 | Safety Evaluation |
| 12.1 | 治験薬が投与された症例数、期間及び用量 | 12.1 | Extent of Exposure |
| 12.2 | 有害事象 | 12.2 | Adverse Events (AEs) |
| 12.2.1 | 有害事象の簡潔な要約 | 12.2.1 | Brief Summary of Adverse Events |
| 12.2.2 | 有害事象の表示 | 12.2.2 | Display of Adverse Events |
| 12.2.3 | 有害事象の分析 | 12.2.3 | Analysis of Adverse Events |
| 12.2.4 | 患者ごとの有害事象の一覧表 | 12.2.4 | Listing of Adverse Events by Patient |
| 12.3 | 死亡、その他の重篤な有害事象及び他の重要な有害事象 | 12.3 | Deaths, Other Serious Adverse Events, and Other Significant Adverse Events |
| 12.3.1 | 死亡、その他の重篤な有害事象及び他の重要な有害事象の一覧表 | 12.3.1 | Listing of Deaths, Other Serious Adverse Events and Other Significant Adverse Events |
| 12.3.1.1 | 死亡 | 12.3.1.1 | Deaths |
| 12.3.1.2 | その他の重篤な有害事象 | 12.3.1.2 | Other Serious Adverse Events |
| 12.3.1.3 | 他の重要な有害事象 | 12.3.1.3 | Other Significant Adverse Events |
| 12.3.2 | 死亡、その他の重篤な有害事象及び他のいくつかの重要な有害事象の叙述 | 12.3.2 | Narratives of Deaths, Other Serious Adverse Events and Certain Other Significant Adverse Events |
| 12.3.3 | 死亡、その他の重篤な有害事象及び他の重要な有害事象の分析及び考察 | 12.3.3 | Analysis and Discussion of Deaths, Other Serious Adverse Events and Other Significant Adverse Events |
| 12.4 | 臨床検査値の評価 | 12.4 | Clinical Laboratory Evaluation |
| 12.4.1 | 患者ごとの個々の臨床検査異常値の一覧表(14.3.4) | 12.4.1 | Listing of Individual Laboratory Measurements by Patient (16.2.8) and Each Abnormal Laboratory Value (14.3.4) |
| 12.4.2 | 各臨床検査項目の評価 | 12.4.2 | Evaluation of Each Laboratory Parameter |
| 12.4.2.1 | 治験期間を通しての臨床検査値 | 12.4.2.1 | Laboratory Values Over Time |
| 12.4.2.2 | 個々の患者の変化 | 12.4.2.2 | Individual Patient Changes |
| 12.4.2.3 | 個々の臨床的に重要な異常 | 12.4.2.3 | Individual Clinically Significant Abnormalities |
| 12.5 | バイタルサイン、身体的所見及び安全性に関連する他の観察項目 | 12.5 | Vital Signs, Physical Findings and Other Observations Related to Safety |
| 12.6 | 安全性の結論 | 12.6 | Safety Conclusions |
| 13 | 考察と全般的結論 | 13 | Discussion and Overall Conclusions |
| 14 | 本文中には含めないが、引用する表、図及びグラフ | 14 | Tables, Figures and Graphs Referred to but Not Included in the Text |
| 14.1 | 人口統計学的データ | 14.1 | Demographic Data |
| 14.2 | 有効性データ | 14.2 | Efficacy Data |
| 14.3 | 安全性データ | 14.3 | Safety Data |
| 14.3.1 | 有害事象の表示 | 14.3.1 | Displays of Adverse Events |
| 14.3.2 | 死亡、その他の重篤な有害事象及び他の重要な有害事象の一覧表 | 14.3.2 | Listings of Deaths, Other Serious and Significant Adverse Events |
| 14.3.3 | 死亡、その他の重篤な有害事象及び他の特に重要な有害事象の叙述 | 14.3.3 | Narratives of Deaths, Other Serious and Certain Other Significant Adverse Events |
| 14.3.4 | 患者ごとの個々の臨床検査異常値の一覧表 | 14.3.4 | Abnormal Laboratory Value Listing (each patient) |
| 15 | 引用文献の一覧表 | 15 | Reference List |
| 16 | 付録 | 16 | Appendices |
| 16.1 | 治験に関する情報 | 16.1 | Study Information |
| 16.1.1 | 治験実施計画書及びその改訂 | 16.1.1 | Protocol and protocol amendments |
| 16.1.2 | 症例記録用紙の見本(内容の異なるページのみ) | 16.1.2 | Sample case report form (unique pages only) |
| 16.1.3 | 治験審査委員会の一覧(確認が行われた年月日、並びに委員の氏名及び職名)、患者への説明文書及び同意書の見本 | 16.1.3 | List of IECs or IRBs (plus the name of the committee Chair if required by the regulatory authority) – Representative written information for patient and sample consent forms |
| 16.1.4 | 治験責任医師及び他の重要な治験参加者の一覧表及び説明(簡潔な(1ページ) 履歴書又は治験の実施に関連する訓練や経験についての履歴書と同等の要約を含む) | 16.1.4 | List and description of investigators and other important participants in the study, including brief (1 page) CVs or equivalent summaries of training and experience relevant to the performance of the clinical study |
| 16.1.5 | 治験総括(調整)医師又は治験依頼者の医学責任者の署名 | 16.1.5 | Signatures of principal or coordinating investigator(s) or sponsor’s responsible medical officer depending on the regulatory authority’s requirement |
| 16.1.6 | 複数のロットが用いられた場合には、治験に用いられたロットごとの薬剤を投与された患者一覧表 | 16.1.6 | Listing of patients receiving test drug(s)/investigational product(s) from specific batches where more than one batch was used |
| 16.1.7 | 無作為化の方法及びコード(患者の識別及び割り付けられた治療) | 16.1.7 | Randomization scheme and codes (patient identification and treatment assigned) |
| 16.1.8 | 監査手順に関する資料、監査証明書(可能であれば) | 16.1.8 | Audit certificates (if available) |
| 16.1.9 | 統計手法に関する文書 | 16.1.9 | Documentation of statistical methods |
| 16.1.10 | 臨床検査に関して施設間の標準化及び品質保証を行ったのであればその方法と手順に関する文書 | 16.1.10 | Documentation of inter-laboratory standardization methods and quality assurance procedures if used |
| 16.1.11 | 治験に基づく公表文献 | 16.1.11 | Publications based on the study |
| 16.1.12 | 総括報告書で引用された重要な公表文献 | 16.1.12 | Important publications referenced in the report |
| 16.2 | 患者データ一覧表 | 16.2 | Patient Data Listings |
| 16.2.1 | 中止症例 | 16.2.1 | Discontinued patients |
| 16.2.2 | 治験実施計画から逸脱した症例 | 16.2.2 | Protocol deviations |
| 16.2.3 | 有効性の解析から除外された症例 | 16.2.3 | Patients excluded from the efficacy analysis |
| 16.2.4 | 人口統計学的データ | 16.2.4 | Demographic data |
| 16.2.5 | 服薬遵守及び(又は)薬物濃度データ(可能であれば) | 16.2.5 | Compliance and/or drug concentration data (if available) |
| 16.2.6 | 個々の有効性反応データ | 16.2.6 | Individual efficacy response data |
| 16.2.7 | 患者ごとの有害事象一覧表 | 16.2.7 | Adverse event listings (each patient) |
| 16.3 | 症例記録 | 16.3 | Case Report Forms |
| 16.3.1 | 死亡、その他の重篤な有害事象発現例及び有害事象による投与中止例の症例記録 | 16.3.1 | CRFs for deaths, other serious adverse events and withdrawals for AE |
| 16.3.2 | 提出された他の症例記録 | 16.3.2 | Other CRFs submitted |





